SCPortalen2
*NEW* - We have added a new Database Discovery Environment, accessible from the side-bar. Here, using common metadata metrics, you can explore publicly available scRNAseq datasets, indexed from NCBI Sequence Read Archive (SRA).
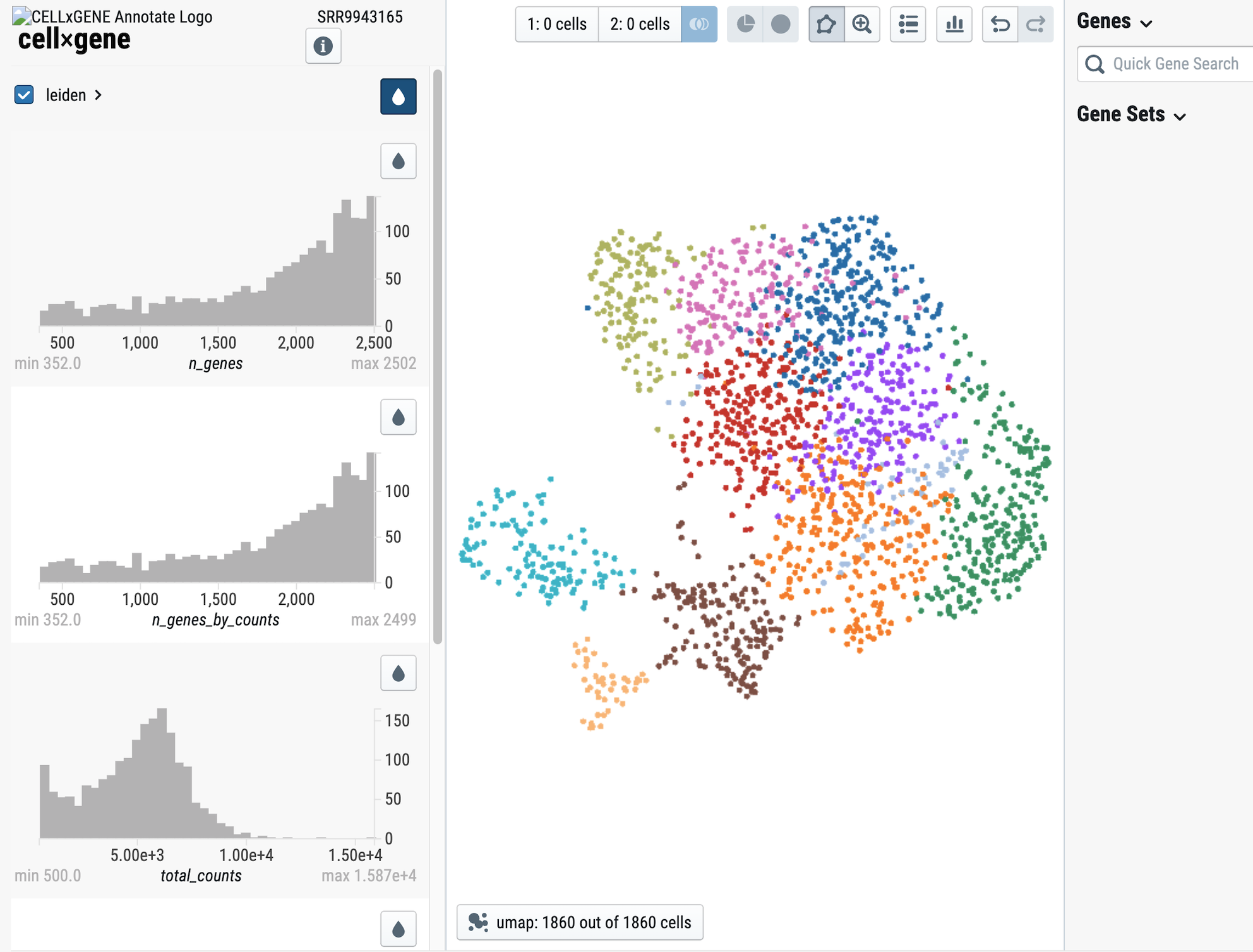 Screen-shot from the dataset GSE135663
Screen-shot from the dataset GSE135663
SCPortalen2: 5' end data, new data visualization and limitless exploration.
We now present SCPortalen2! But what's changed? In this new version we've made some changes to improve the quantity and quality of data as well as a new service to explore thousands of scRNA-seq datasets. We're expanding the reach of SCPortalen by processing 5' end data with a brand new pipeline, based on FANTOM5 specifications. SCPortalen2 will become the first stop for reseachers looking into single-cell promoter/enhancer activity so we provide TSS tag counts as a starting point for downstream analyses. For data analyzed with 10x Genomics' protocols, we provide a new interactive UMAP plot with the aid of the CellxGene tool. Finally, as already announced we have released the Single-cell Dataset Discovery (SCDD) interface, we've indexed every publicly available human and mouse scRNA-seq dataset from NCBI SRA . Follow the link in the sidebar and start exploring!
A resource for the single-cell research community-
Single-cell omics recently emerged as powerful tools to investigate heterogeneity of large populations of cells. Among others, improvements in sequencing, microscopy and microfluidic technologies let to a rapid increase of complex datasets with single-cell resolution. However, due to a lack of available platforms to easily share and integrate complex single-cell datasets, the accessibility of such datasets can be a barrier to efficient usage.
To address the issue of efficient single-cell data management, we introduced a single-cell data integration platform. The motivation for the development of this database was to enable easy access, integration and collaboration on single-cell datasets generated by the research community worldwide. This database features integration of single-cell metadata, cell images and sequence information. Currently the platform is divided into two parts; the first part is the single-cell dataset generated at the Division of Genomic Technologies at RIKEN CLST and the second part is the single-cell dataset published in peer-reviewed journals.
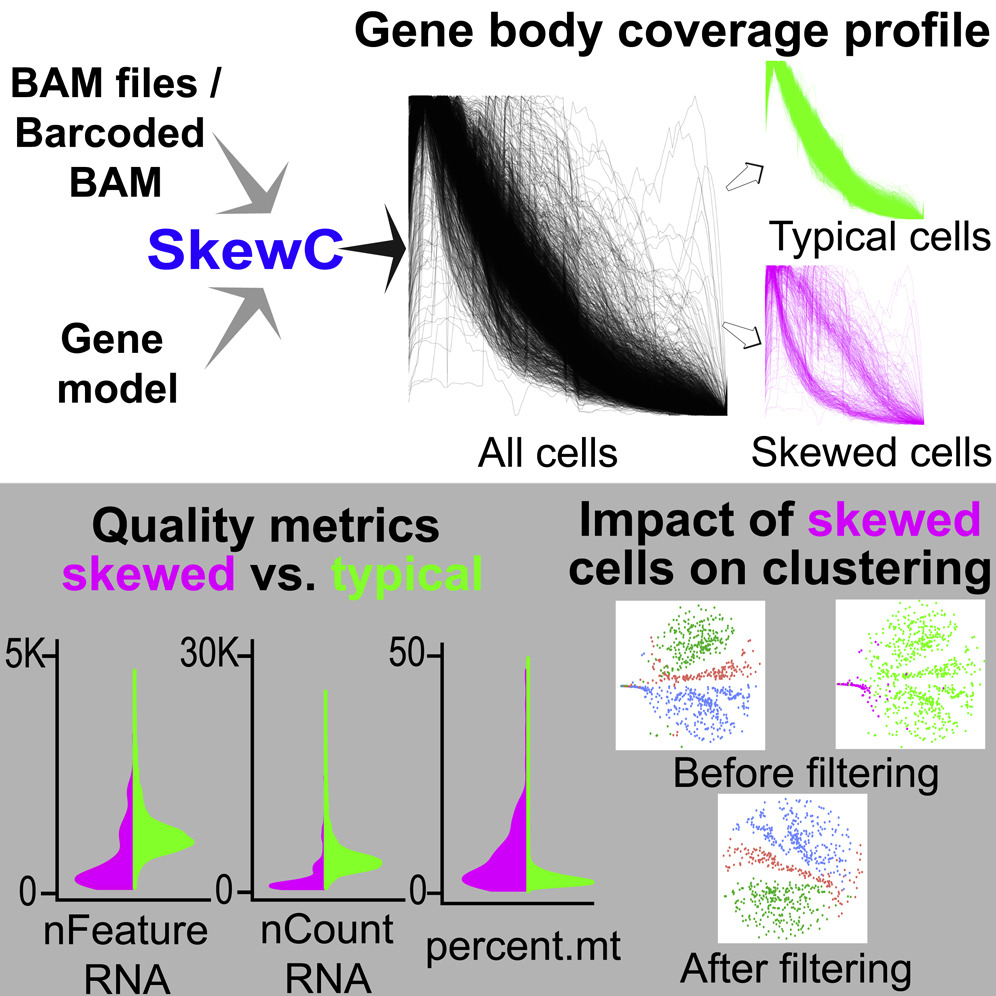 Graphical abstract for the SkewC paper. DOI:doi.org/10.1016/j.isci.2022.103777
Graphical abstract for the SkewC paper. DOI:doi.org/10.1016/j.isci.2022.103777
SkewC: Identifying cells with skewed gene body coverage in single-cell RNA sequencing data
Researchers at LSBDT have created the SkewC QC tool. The methodology is based on the assessment of gene coverage for each cell, and its skewness as a quality measure. SkewC is capable of processing any type of scRNA-seq dataset, regardless of the protocol. This tool is designed to avoid misclustering or false clusters by identifying, isolating, and removing cells with skewed gene body coverage profiles.